今天介绍一种新的细胞分割方法Proseg,在细胞边缘分割处理上更加精确,从而满足空间转录组学分析的要求。
文章标题为《Cell simulation as cell segmentation》,2025年5月22发表在Nature Methods上。
空间转录组学中需要把单细胞的区域和转录信息对应起来,然而这也对细胞边缘部分的分割精度提出了更高的要求,否则一不小心转录信息就被映射到相邻的细胞或者空白的位置。
Many years of development have gone into refining image-based cell segmentation, which long predates in situ spatial transcriptomics. Early success was found using heuristics such as the watershed algorithm that operate on single-channel images. More recently, the task has fallen to deep-learning models such as U-Net, Mesmer and Cellpose. As in all deep-learning image-recognition tasks, the model is only successful to the extent that it was trained on representative data. In the cell segmentation task, this can be problematic, both because cells come in all shapes and sizes, and because different platforms and experiments may use differing staining strategies. Lacking a pre-trained model, a practitioner is forced to either manually label data to train their own, or risk mis-segmentation with a potentially ill-fitting model. Though recent work has focused on trying to improve how well models generalize, this remains a major hurdle.
细胞分割事实上已经有非常多的好的方法,但实际上研究者拿到的数据总是充满个性化(细胞形态的多样化,以及染色方法的些微差别都会导致这种个性化差异),这就意味着缺少特别好的预训练模型,最终的分割精度无法保证。在对精度要求很高的场景下,就不得不重新标注训练。。。当然也可以利用很多很多数据训练一个更加general的模型,但是这条路也是相当困难。所以这个时候就得另辟蹊径了。这里我们先直接看下效果:
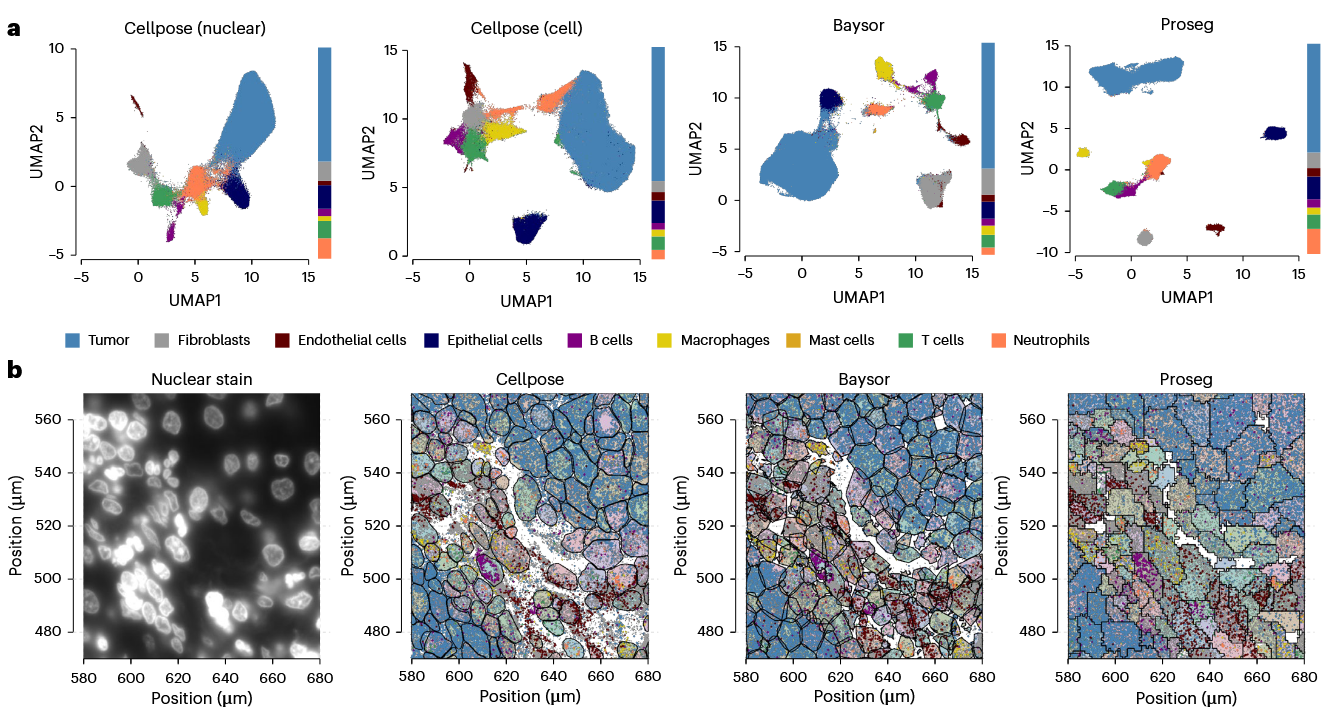
这里比较了cellpose, Baysor 和 Proseg进行单细胞分割的效果,可以看到cellpose分割完之后,单细胞转录组的数据(a)分开得不是很好,而Proseg效果是最好的。从下面的细胞图像和空间转录组的mapping图像来看,Proseg寻找到的边界严格意义上讲并不是单细胞边界(可能多个细胞合并),而是单类细胞边界。因为Proseg的边界不依赖于细胞染色图像,而是直接从空间转录组学数据入手的,所以我认为Proseg本质上是一种聚类方法。
The method takes as its inspiration Cellular Potts models (CPMs), which simulate rudimentary cellular behaviors on a lattice of pixels or voxels by optimizing a contrived energy function. With Proseg we employ a similar strategy to generate cell morphologies, but in place of a contrived function we use a probabilistic model of the spatial distribution of observed transcripts. Cell morphologies are initialized using a nuclear stain, then expanded and altered at random until they best explain the observed data.
但文章说的是通过模拟实现的,借鉴的是1992年的一个工作提出的方法,使用像素代表细胞区域,然后通过设计一个函数来定义细胞模拟的行为,我们进一步看看是怎么回事。
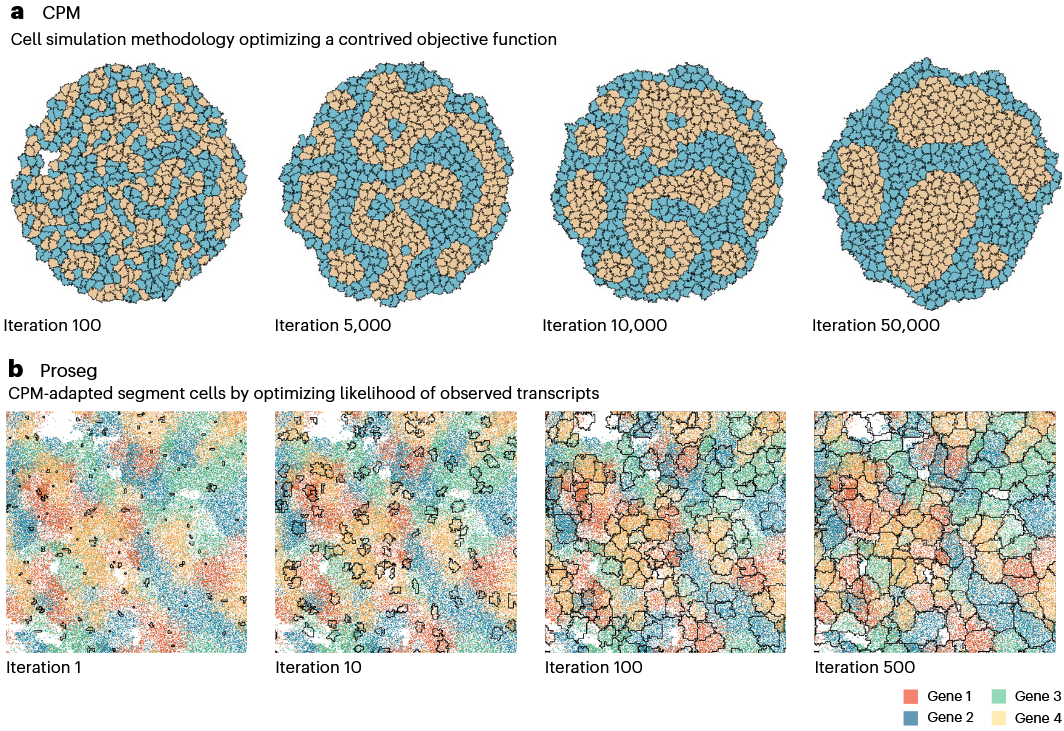
上图第一行就是细胞形态模拟CPM方法的一个示例,函数定义了相似的细胞倾向于靠在一起,通过多次迭代后,确实就形成了若干cluster。当然你也可以设计更加复杂的函数,把能量以及其它各种相互作用考虑进去,那么结果会更复杂。而Proseg先根据细胞核染色成像结果生成种子,然后模拟细胞边缘得变化。具体细胞行为函数定义是,提供的四种基因的转录组都是高度cell specific,所以寻找到的细胞边界要尽可能地使一个细胞区域内单个基因出现的频率最高。
所以这里相当于是使用 highly cell specific 的 gene 完成了某种细胞类型的“标记染色”,然后通过Proseg得到一个概率细胞分割区域,然后再mapping到其它感兴趣基因的转录组上。
这样做感觉有一些明显的弊端。比如找到这个highly cell specific gene就比较难找到合适的,因为它不仅得是细胞特异性表达,还得表达得多,否则Proseg计算边界时估计错误率也挺高。另外由于Proseg找到的细胞边界,有可能将同类多个细胞融合,这也会导致后续进行一些单细胞的定量分析时的不准确问题。
最后看看审稿人都是啥意见吧。

果然,2号审稿人指出的问题比较致命,由于该方法过于简化的假设和测试,这个方法在实际复杂数据上的表现被质疑。然后作者用神经元的数据进行了更丰富的测试。


此处评论已关闭